Free radical intermediates are generally involved in reactions carried out at high temperatures, under the influence of light or in the presence of free radical generators. Even at room temperature, many reactions proceed by a free-radical mechanism, particularly in nonpolar solvents and in the presence of a radical producer. A radical has a choice: it can find another radical and combine to form a spin-paired molecule, or it can react with a spin-paired molecule to form a new radical; both are possible. A third alternative is to decompose in a unimolecular reaction, giving rise to a new radical and a spin-paired molecule. A fourth class of reaction, rearrangement, is relatively rare. Most common radicals are extremely reactive and are difficult to isolate. Such radicals are referred to as short-lived radicals in contrast to stable, free radicals that are not so reactive and exist in equilibrium with the normal compounds.
Radical reactions are frequently found to occur as chain reactions composed of three types of processes:
- an initiation step (for example, one of the generation reactions discussed in the previous section),
- a series of propagation steps, and
- one or more termination steps that stop the chain reaction.
The following are some characteristics of free-radical reactions:
- Reactions are almost similar whether they are taking place in the vapour or liquid phase, though solvation of free radicals in solution does cause some differences.
- Acids, bases and polar solvents have almost no effect on radical-reactions.
- Free-radical initiators are necessary.
- The rates of free-radical reactions are decreased or suppressed by inhibitors, for example, NO, O2, benzoquinone, etc.
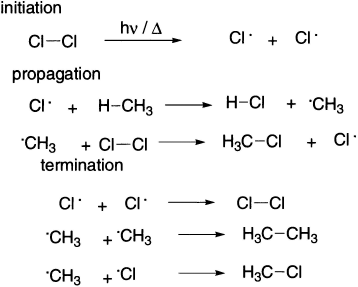
Each propagation step in a radical chain involves the reaction of a species with one unpaired electron to produce another species having an unpaired electron. Moreover, the reactant in one step is a product in a subsequent step. One example of a propagation step in a radical chain reaction is the abstraction of a hydrogen atom by a halogen in free radical halogenations. Methane and chlorine do not react in the dark at room temperature, but under the influence of UV light or at temperature of 250–400°C reacts vigorously to yield HCl and a mixture of CH3Cl, CH2Cl2, CHCl3 and CCl4.
The order of reactivity of the halogens for methane can be explained by energy considerations. ΔH values for the two principal propagation steps are:
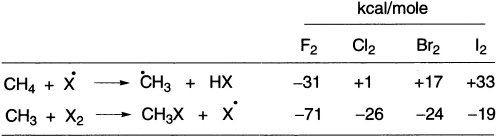
The corresponding reaction with iodine atoms is so strongly endothermic that the reaction is ineffective; indeed, HI can reduce alkyl iodides to alkanes and iodine. For fluorine, both propagating steps are strongly exothermic; reaction is violent and is accompanied by the fragmentation of alkyl groups.
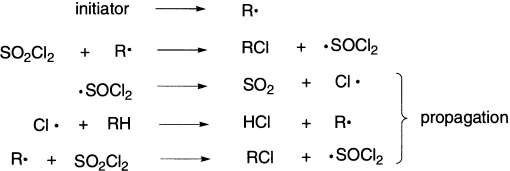
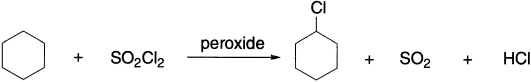
Radical-catalyzed chlorination and bromination occur readily on alkanes and substituted alkanes, both in the gas phase and in solution. Both thermal and photochemical generations of the halogen atoms are employed, and for chlorinations, sulphuryl chloride in the presence of an initiator such as dibenzoyl peroxide is also used.
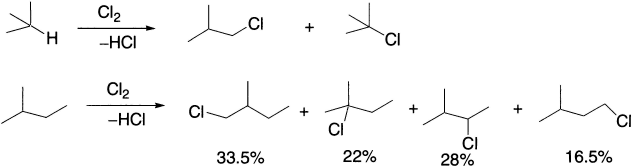
For example, the chlorination of isobutene in the gas phase at 100°C gives comparable quantities of isobutyl chloride and t-butyl chloride, and at 300°C 2-methyl butane gives the following products:
Cyclohexyl chloride can be isolated in 89% yield by treating cyclohexane with sulphuryl chloride in the presence of dibenzoyl peroxide.
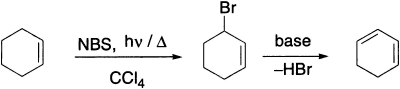
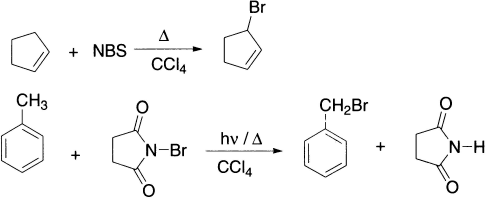
N-bromosuccinimide (NBS) is a very specific reagent for allylic and benzylic bromination. NBS acts as a bromine reservoir, maintaining a low concentration of molecular bromine by reacting with HBr. The bromine molecule dissociates into bromine atoms in the presence of light or radical initiators. H-abstraction by bromine atom generates an allylic radical which then reacts with the Br2 to yield the product. Allylic compounds of the type RCH=CH-CH2R’ give mixtures of two monohalogenated products because the allylic radical can react at each of the two carbon atoms.
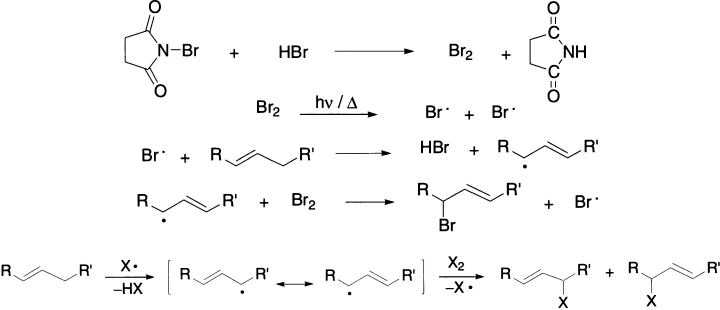
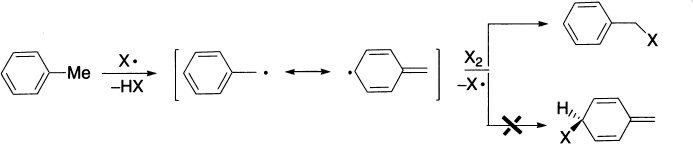
Benzylic compounds, on the other hand, give only one product because the possible isomers, being non-aromatic, are of much higher energy content.
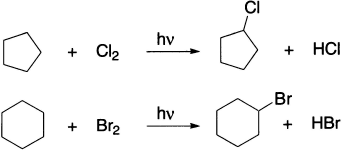
Cyclic compounds undergo the same reactions as do noncyclic compounds. For example, cyclic alkanes and cyclic alkenes undergo radical substitution reactions with chlorine or bromine.
The bromination of allylic compounds is generally used in the conversion of alkenes into conjugated dienes, which are obtained from the allylic bromides by base-catalyzed elimination.
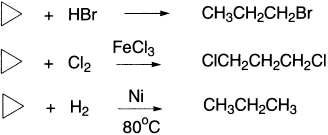
Cyclopropane is one notable exception, which undergoes electrophilic addition reactions. Cyclopropane is more reactive than propene toward addition of acids such as HBr and HCl but is less reactive toward addition of Cl2 and Br2, so a Lewis acid, FeCl3, is needed to catalyze halogen addition.
A carbon-centred radical can also abstract an atom from another molecule (or from another atom in the same molecule) to fill its outer shell if the process is radical trapping, in which a radical abstracts a hydrogen atom from a facile hydrogen atom donor such as tri-n-butyltin hydride (Bu3SnH). This reaction can serve as a means of detecting radical intermediates, because the appearance of the species R–H upon addition of Bu3SnH to a reaction suggests the intermediacy of R. in the reaction.
H3C· + HSn(Bu)3 → CH4 + ·Sn(Bu)3
In some cases, the acceptor is usually a transition metal ion in a high-valence state, such as hexacyanoferrate(III) ion in the oxidation of certain phenols:
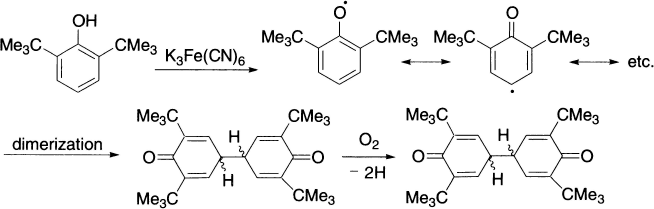
The pinacol reaction is an example of radical dimerization.
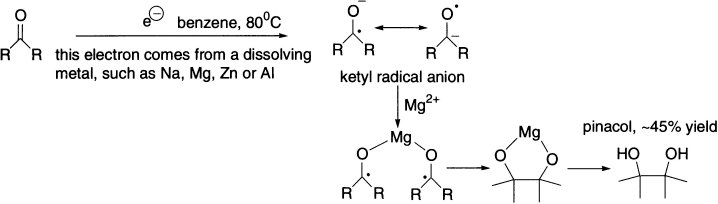
Trialkyltin hydrides are effective hydrogen atom transfer agents, and trialkyltin radicals will abstract chlorine, bromine, or iodine atoms from alkyl halides. Together with a radical initiator such as azobisisobutyronitrile (AIBN), trialkyltin hydrides and alkyl halides can give reduction or other radical-derived products.
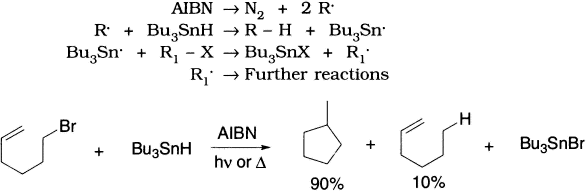
Another common propagation step is addition of a radical to a double or triple bond, as in the anti-Markovnikov addition of HBr to an alkene.
Carbon tetrachloride can be added to propylene in 80% yield.
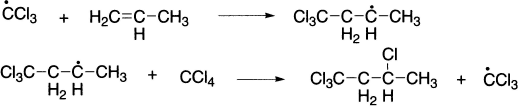
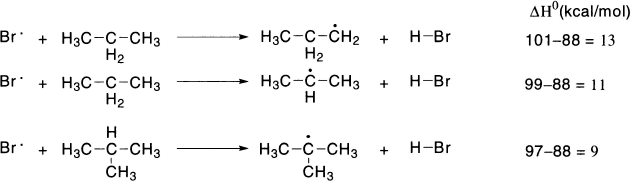
Relative rates of alkyl radical formation by a chlorine radical is tertiary (5.0) > secondary (3.8) > primary (1.0), while relative rates by a bromine radical is tertiary (1600) > secondary (82) > primary (1).
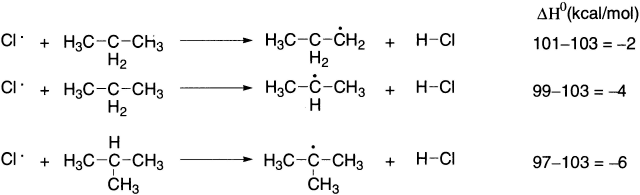
When bromine radical is the hydrogen-abstracting agent, the differences in reactivity are very great. To answer this question, we must look at the ΔH0 values for formation of primary, secondary and tertiary radicals as a result of reaction with chlorine and bromine radicals. These ΔH0 values can be calculated using the bond dissociation energies.
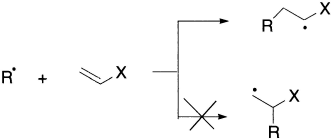
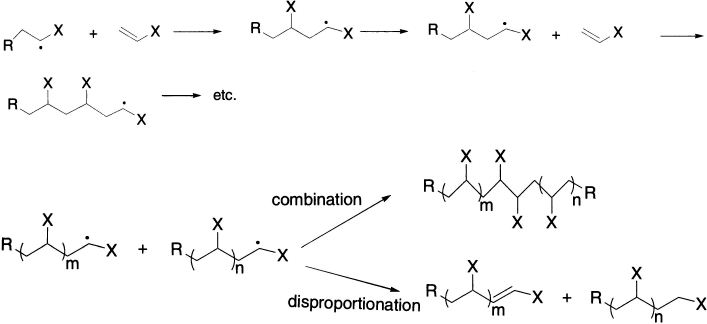
Radical addition to a multiple bond is the key step in radical polymerization. For example, in the polymerization of styrene, the initiation step is homolysis of an initiator, which produces a radical that adds to a styrene molecule to begin the polymer chain. Two carbon–carbon single bonds are more stable than one carbon–carbon double bond by about 20 kcal/mol, so the difference in the strengths of carbon–carbon single and double bonds provides the driving force for the reaction. Increasingly, radical addition reactions are finding application in organic synthesis. Because charged species are not involved in the reaction, subtle effects due to orbital interactions and steric interactions can provide opportunities for stereoselective syntheses.
Another useful chain reaction involves the PTOC (pyridine-2-thione-N-oxycarbonyl) esters developed by Barton. Reaction of a carboxylic acid chloride (RCOCl) with the sodium salt on N-hydroxypyridine-2-thione produces an ester designated as R–PTOC. Addition of radical Y· (formed by an earlier initiation step) to the R-PTOC leads to the carboxy radical RCO2·. The carboxy radical then decarboxylates to produce the radical R·, which can continue the chain reaction or can undergo other reactions.
Another reaction of radicals is rearrangement. Radicals are generally less susceptible to rearrangement than are carbocations, and the 1,2 hydrogen or carbon shifts seen with carbocations are not observed with radicals. However, apparent phenyl migration has been observed. Treatment of neophyl chloride with phenylmagnesium bromide and cobaltous chloride produced isobutylbenzene (15%), 2-methyl-3-phenyl-1-propene (9%) and β,β-dimethylphenylethyl, (25%). The 1,1-dimethylspiro [2,5] octadienyl radical has been proposed as an intermediate or transition structure in the rearrangement.

Rearrangements can also occur when they result in the relief of strain in a cyclic system, as in the radical-catalyzed addition of carbon tetrachloride to β-pinene.
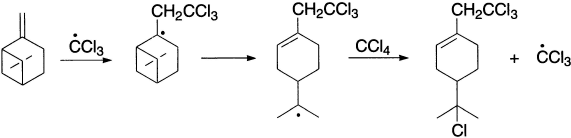
Migrations have also been observed for chloro groups. Migration of a halogen could occur via a transition state in which the odd electron is accommodated in a vacant d-orbital of the halogen.

The characteristic of radical termination steps is that two radical centres react to produce a product, each of which has an even number of electrons. The two most important processes are dimerization and disproportionation. Dimerization is the reverse of the thermal dissociation of a σ bond to produce two radicals. This process is usually quite exothermic and has small or negligible activation energy. However, significant steric hindrance can lower the dissociation energy and introduce a barrier to recombination. For example, the dissociation energy of 1-(2,6-dimethylphenyl)-2-(2,6-dimethylphenyl) ethane is only 22 kcal/mol.
H3C· + ·CH3 → H3C − CH3
Disproportionation can be considered to be hydrogen abstraction from another radical. The overall result is the destruction of two radicals, one radical centre being converted to a carbon–hydrogen bond, the other being converted into a double bond. Both dimerization and disproportionation represent bimolecular processes of radicals. Dimerization is more exothermic than is disproportionation, but the ΔS for dimerization is much more negative than is ΔS for disproportionation. Therefore, the reaction of two radicals depends strongly on temperature: higher temperature favouring disproportionation, lower temperature favouring dimerization.

There is one important exception to the statement that radical termination steps produce products with an even number of electrons. A radical addition step may produce a radical product that is much less reactive than the reacting precursor; so further addition may be precluded. This process, which is known as spin trapping, is primarily useful as a means of studying radicals that cannot be studied directly by EPR. Adding a nitroso compound or a nitrone to a reaction mixture involving short-lived radicals can produce a spin adduct, a longer-lived species that can be studied directly by EPR spectrometry. The spectrum of the product is often diagnostic of its radical precursor.
Both alkyl and aryl radicals substitute in aromatic nuclei by an addition–elimination sequence. Substituents in the orth– and para-positions are able to increase the delocalization of the intermediate radical and consequently activate these positions to substitution.
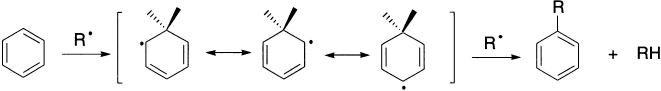
The coupling of two aromatic rings cannot be explained on the basis of simple abstraction. The products can be explained by a mechanism similar to that of electrophilic and nucleophilic aromatic substitution. In the first step, the radical attacks the ring; the intermediate formed is stable due to resonance. The reaction may terminate in three ways: by simple coupling, by disproportionation, or simply by hydrogen abstraction.
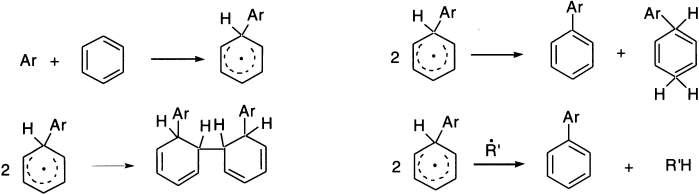
All substituents (electron-donating and electron-withdrawing) increase reactivity at ortho– and para-positions over that of benzene, at meta-position reactivity is usually similar to that of benzene.
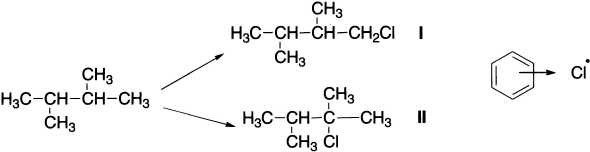
A solvent usually has little effect on free-radical substitutions, however, in certain cases, it can make an appreciable difference. The chlorination of 2,3-dimethylbutane in aliphatic solvents gave about 60% I and 40% II, but in aromatic solvents, the ratio became about 10:90. This result is attributed to complex formation between the aromatic solvent and the chlorine atom, which makes the chlorine less reactive and more selective.
Free-radical reactions may be divided into two classes. In the first, the product results from the combination of two radicals, as in the Kolbe synthesis. Electrolysis of the alkali metal salts of aliphatic carboxylic acids results in the liberation of alkyl radicals at the anode and their subsequent dimerization.
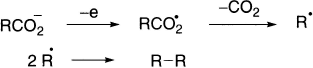
In the second class, the product results from the reaction of a radical with a molecule, as in the case of photochemical chlorination of methane. The fundamental difference between the two types of reactions is that the latter class involves chain reactions.
Leave a Reply