In most of the cationic shifts described so far, a hydrogen atom migrates with a pair of bonding electrons (hydride migration). However, alkyl groups also can shift (along with the electrons from the σ bond that connects the group to the adjacent atom). It was George Wagner who observed in 1899 that terpenes undergo skeletal reorganizations. Hans Meerwein provided the second key to the conceptual puzzle in 1922. Later, in 1927, Meerwein generalized the carbocationic mechanism to account for other terpenic rearrangements.
When an alkyl group migrates to a cationic centre, there are changes in the carbon skeleton, and the reaction is referred to as a Wagner–Meerwein rearrangement Alkyl migration occurs in order to make a carbocation more stable; thus, the driving force of the rearrangement is the formation of a carbocation with greater stability. It is a [1,2] rearrangement of H atoms or alkyl group in carbenium ion that is generated in one or more steps, and the rearranged carbenium ion reacts further in one or several steps to give a valence-saturated compound. The rearrangement is catalyzed by Lewis acids like AlCl3, FeCl3, TiCl4 and SnCl4 or by Bronsted acids and is stereospecific. The migrating group approaches the electron-deficient carbon from the direction opposite to that of the leaving group, that is, the SN2 type geometry is needed. The reaction proceeds faster in polar solvents and is consistent with a first order rate law. The order observed for some solvents is SO2 > MeNO2 > MeCN > PhOMe > PhBr > PhH > Et2O. The classical examples are the transformation of camphenilol to santene and camphene hydrochloride to isobornyl chloride. Isobornyl chloride, the exo-isomer (that is, the chlorine atom on the side opposite to the migrating bridge), is the sole product, which slowly rearranges to the thermodynamically more stable endo-isomer (bornyl chloride). Bornyl chloride is also obtained by treatment of α-pinene with HCl.
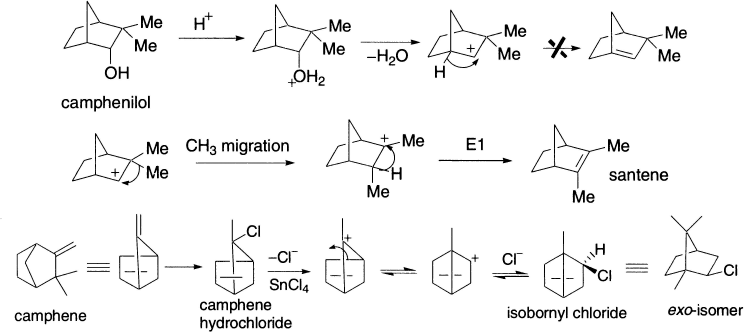
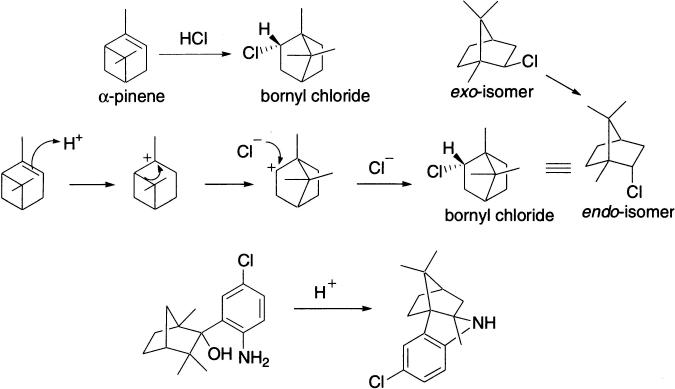
Another point of interest in the above reaction is reversibility. The migration terminus in the forward process becomes migration origin in the reverse process. It, therefore, follows from the principle of microscopic reversibility that both reactions should have a common reaction mechanism and identical stereochemical rules. One can summarize these rules in the following way. These stereochemical rules have a general applicability for all 1, 2-shifts.

The carbocation may be generated in a variety of ways.
- From a halide, by using a strongly ionizing solvent or by adding a Lewis acid such as silver ion.
- By treatment of an alcohol with acid.
- From an amine by treatment with nitrous acid.
- From an alkene by protonation.
In the trans-decalin derivative (depicted after this para), the hydroxyl group is held in the equatorial position because the ring system cannot flip. In this situation, there is no hydrogen anti to the hydroxyl, but two of the ring carbon atoms are in the appropriate anti position for rearrangement (that migrates leaving the more stable carbocation) and, in the presence of acid, ring contraction takes place.

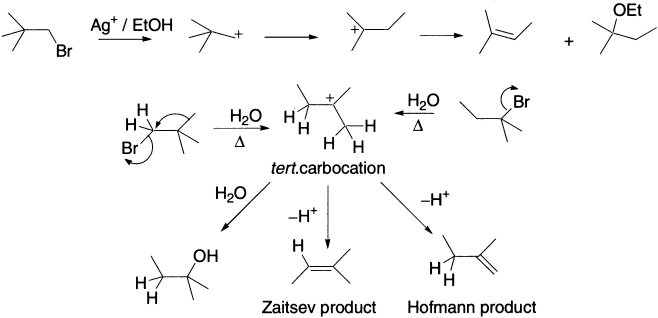
Wagner–Meerwein rearrangements of cations are similar in detail to those in which hydrogen atoms migrate: for example, the solvolysis of 1-bromo-2,2-dimethylpropane. Heterolytic cleavage of the C-Br bond of 1-bromo-2,2-dimethylpropane leads not to the primary cation but to the more stable tertiary cation. This cation is produced when a methyl group migrates from C-2 to C-1 as the C-Br bond is broken. The simultaneous migration of the alkyl group and departure of the leaving group to form a tertiary cation is, therefore, faster than the simple loss of the leaving group to form a primary cation.

The products observed are those that result from further reaction of rearranged (tertiary) cation. The alcohol results from reaction of the tertiary cation with water, forming an oxonium ion. Loss of a proton generates the product alcohol with a rearranged carbon skeleton. Alternatively, cation can lose a proton from either of two different adjacent sites to give either the Zaitsev or Hofmann elimination product. All three observed products derive from the rearranged cation, whose carbon skeleton differs from that of the starting material because of migration of methyl group from C-2 to C-1 in a Wagner–Meerwein rearrangement.
When a more stable intermediate can be formed by migration of an alkyl group or hydrogen, rearrangement nearly always occurs. The products formed depend on the structure of the intermediate cation, no matter how this cation is initially formed. For example, the products obtained from the solvolysis of 2-bromo-2-methylbutane are the same as those from the solvolysis of 1-bromo-2,2-dimethylpropane. When a driving force for cation rearrangement exists, migration of an alkyl group almost always takes place faster than trapping of a less stable cation by solvent or another nucleophile.
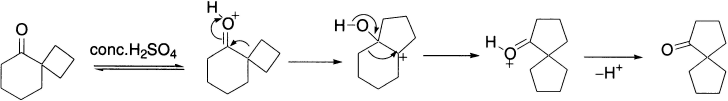
Ring Expansion. Cation rearrangements can be driven thermodynamically by factors other than the degree of substitution of the cation. For instance, ring strain is also important. As an example, let us consider the treatment of cyclobutylmethanol with strong acid:

Protonation of the hydroxyl oxygen forms an oxonium ion that loses a water molecule, generating a primary cation. The carbinol carbon is next to a strained four-member ring; an adjacent methylene group (CH2) migrates to this centre with the electrons of the C-C σ bond and, at the same time, water is also lost. Both a reduction in strain (a four-member ring becoming a five-member ring) and an increase in the degree of substitution (from a primary to a secondary cation) are accomplished by this migration. The resulting cyclopentyl cation is then captured in a slower step by an external nucleophile. When treated with aqueous, HBr cyclobutylmethanol is converted to cyclopentyl bromide.
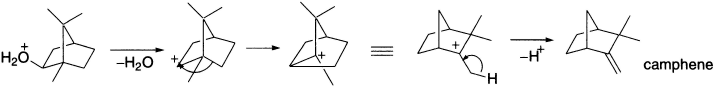
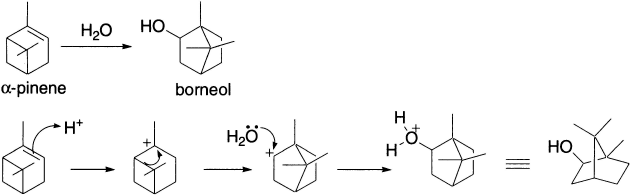
In directing the course of the reaction, relief of ring strain can sometimes be more important than the degree of substitution of the carbocationic centre. Consider, for example, the acid-catalyzed solvolysis of α-pinene. Recall that cations can be produced by protonation of alkenes (the first step in electrophilic addition). Protonation of α-pinene forms the tertiary carbocation, which is favoured over the alternative carbocation (recall Markovnikov’s rule). Though the rearrangement step transforms a stable tertiary cation into a less stable secondary cation, relief of strain in expansion from a four- to a five-member ring makes the alkyl migration favourable. The secondary cation is then trapped by water, ultimately producing a product alcohol with a carbon skeleton different from that of the starting material.
A carboxonium ion may become less stable than a carbenium ion only when ring-strain effects dominate. In such cases, carbenium ions can be generated from carboxonium ions by way of a Wagner–Meerwein rearrangement. Thus, the decrease of ring strain can provide a driving force strong enough to overcompensate for the conversion of a more stable into a less stable cationic centre.
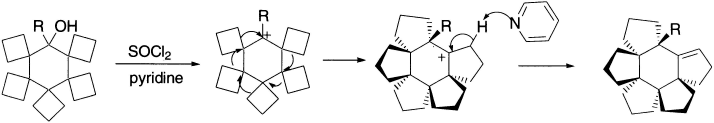
A tandem rearrangement and a cascade rearrangement describe sequences of two or more rearrangements taking place more or less directly, one after the other. The following example involves five [1,2]-rearrangements, each one effecting the conversion of a spiro-annulated cyclobutane into a fused cyclopentane.
Every polycyclic hydrocarbon having the molecular formula C10H16 can be isomerized to adamantane. Adamantane, possessing the structure of the repeating unit of the diamond lattice, is the stablest alkane of molecular formula C10H16. All other isomeric tricyclic alkanes are converted to adamantane if subjected to sufficiently vigorous Lewis acid treatment. The minimum number of [1,2]-rearrangements needed in such rearrangements is so high that it can be determined only with the use of a computer program. The rearrangements occur in the presence of catalytic amounts of AlCl3 and tert-BuCl. These isomerizations almost certainly involve [1,2)-shifts of H atoms as well as of alkyl groups. One cannot exclude that [1,3]-rearrangements may also play a role.
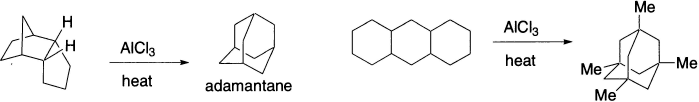
A novel application of this rearrangement is the formation of adamantane in 15% yield by treatment of the reduction product of the readily available dimer of cyclopentadiene with AlCl3 at 150–180°C. The reaction product, adamantane, is formed under thermodynamic control. It is the so-called stabilomer (the most stable isomer) of all the saturated hydrocarbons having the molecular formula C10H16.
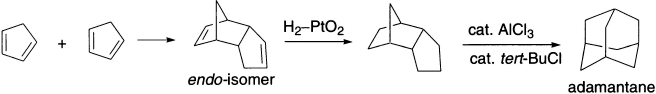
This impressive cascade reaction begins with the formation of a small amount of the tert-butylcation by reaction of AlCl3 with tert-BuCI. The tert-butylcation abstracts a hydride ion from the substrate C10H16. Thus, a carbenium ion with formula C10H15+ is formed. These carbenium ions C10H15+ are certainly substrates for Wagner–Meerwein rearrangements and also potential substrates for [1,3]-rearrangements, thereby providing various isomeric cations iso-C10H15+. Some of these cations can abstract a hydride ion from the neutral starting material C10H16. The saturated hydrocarbons iso-C10H16 obtained in this way are isomers of the original starting material C10H16. Such hydride transfer, and [1,2]- and [1,3]-shifts, respectively, are repeated until the reaction arrives at adamantane by way of the adamantyl cation.
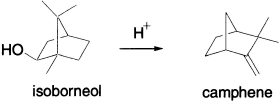
1,2-Methyl shifts in terpenes: the Nametkin rearrangement This particular type of Wagner–Meerwein shift has special recognition due to its importance in the field of terpene chemistry. For example, the conversion of α-methyl camphene to 4-methylisoborneol involves both a Nametkin and a Wagner–Meerwein rearrangement.

Nametkin rearrangements are commonly encountered in acid-catalyzed dehydrations of 3,3-dimethyl[2.2.1]bicycloheptan-2-ols such as in the camphenilol derivatives shown, and these can be seen to be further examples of rearrangements of neopentyl derivatives.

Leave a Reply