Stevens Rearrangement
In the Stevens rearrangement, keto-quaternary ammonium or tertiary sulphonium salts rearrange to amino ketones or mercapto ketones under the influence of strong base. Quaternary ammonium ions, which contain ß-hydrogen atoms undergo E2 (Hofmann) elimination with base:
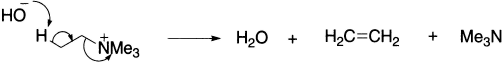
If, however, none of the alkyl groups possesses a ß-hydrogen atom, but one has a ß-carbonyl group, an α-hydrogen is removed by base to give an ylide followed by rearrangement. The rearrangement does not proceed readily when the benzoyl group is replaced by a phenyl or alkyl group, these being relatively ineffective in ‘labelizing’ the hydrogens on an adjacent methylene group. The reaction is accelerated by base, when slightly more than one equivalent is added.
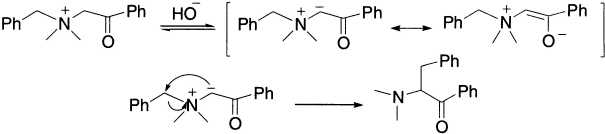
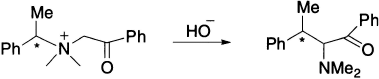
When the rearrangement is carried out on the optically active ammonium ion, the migrating group migrates with retention of configuration. The migrating group may be of the types allyl, benzhydryl (Ph2CH−) or phenacyl (PhCOCH2−).
When various substituents are placed in the meta– or para– position of the benzyl group, the reaction rate is increased in the order of electron-attracting ability of the substituent. In contrast, para-substituent in the phenacyl group increases the rate of rearrangement with the order of their electron-releasing ability. The rearrangement is retarded by incorporation of electron-attracting substituents in the phenacyl group because of the lower electron density at the negatively charged attacking carbon. The rearrangement is also retarded by incorporation of electron-attracting substituents into the benzoyl group.
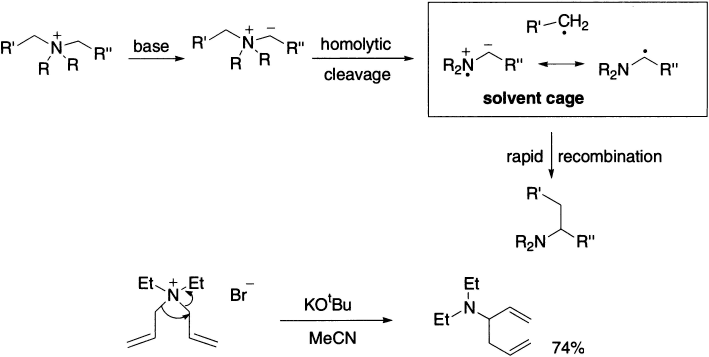
The rearrangement occurs in a concerted manner, probably via formation and combination of radical-pairs.
The methylene group of a propargyl moiety is similar in chemical behaviour to an active methylene group adjacent to a carbonyl group because of the acidity of α-carbon atom to the acetylenic function. The propargyl group has been shown to be capable of acting as both the migrating group and the migration terminus in the Stevens rearrangement, that is, it forms a carbanion and promotes the cleavage of C-N bond. In the following example, the propargyl group acts as a migrating group and the phenacyl group as a migration terminus.
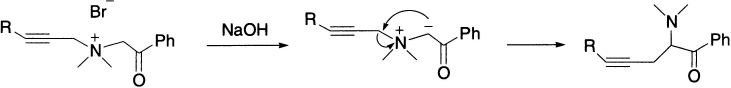
Here, the benzyl group acts as a migrating group and the propargyl group as a migration terminus.
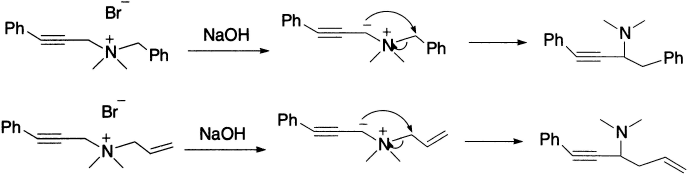
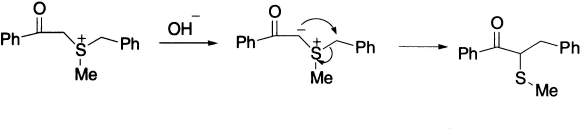
Similar rearrangements have been observed with sulphonium salts under sufficiently drastic conditions. For example, benzyl methyl phenacyl sulphonium ion gives α-methyl mercapto-α-benzyl acetophenone.
Wittig Rearrangement
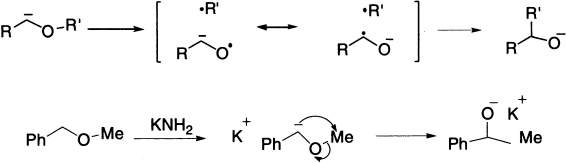
The Wittig rearrangement is directly analogous to the Stevens rearrangement in mechanism and outcome (not to be confused with the better known Wittig reaction) and takes its name from George Wittig who postulated the mechanism of the reaction. It is an example of [2,3] sigmatropic rearrangement in which benzyl and allyl ethers undergo a base-catalyzed rearrangement to α-allyl alkoxides, analogous to the Stevens rearrangement. A benzylic or allylic carbanion is generated by the action of a powerful base such as amide ion or phenyl lithium, and migration of carbon then leads to the more stable oxyanion. The stereochemistry of the Wittig rearrangement can be predicted in terms of a cyclic five-member transition state in which the α substituent prefers an equatorial orientation. Migratory aptitudes are in the order allyl ~ benzyl > alkyl > methyl > aryl, with electron-withdrawing substituents increasing aryl group migratory aptitude.
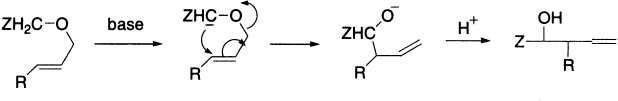
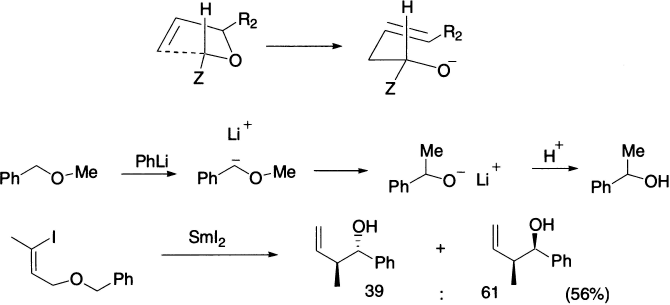
As with the Stevens rearrangement, there is evidence for a radical-pair mechanism:
[2,3] Sigmatropic rearrangements of anions of N-allyl amines have also been observed and are known as aza-Wittig rearrangements. The reaction requires anion-stabilizing substituents and is favoured by N-benzyl and by silyl or sulphenyl substituents on the allyl group.
Favorskii Rearrangement
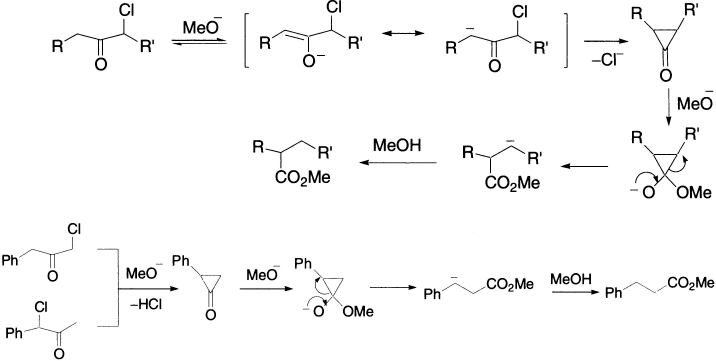
α–Halo ketones react with base to give enolates that rearrange to esters via cyclopropanones. The direction of ring opening of the cyclopropanone is determined by which is more stable of the two possible carbanions that can be formed. Alkyl groups destabilize carbanions, whereas aryl groups stabilize them by delocalization of the negative charge. For example, PhCH2−COCH2Cl and PhCHCl−COMe give the same product. Chloroketones are normally preferable to bromoketones as reactants in the Favorskii rearrangement. It can be classified as intramolecular rearrangement from carbon to carbon, involving a migrating group moving without its electron from migrating origin to an electron–rich terminus.
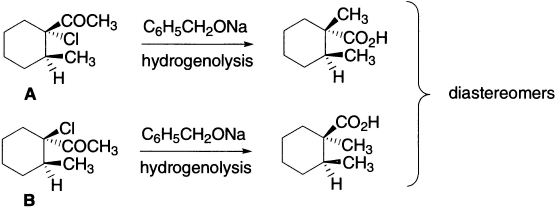
A clear cut case of stereospecific rearrangement has been demonstrated using a pair of epimeric 1-chloro-1-acetyl-2-methylcyclohexanes A and B of proven configuration.
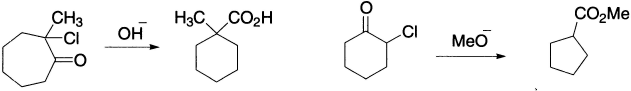
The rearrangement can be employed for bringing about ring contraction in cyclic systems, for example, 2-chlorocyclohexanone and methoxide ion give methyl cyclopentanecarboxylate in 60% yield.
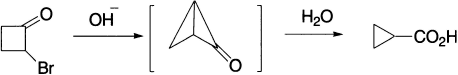
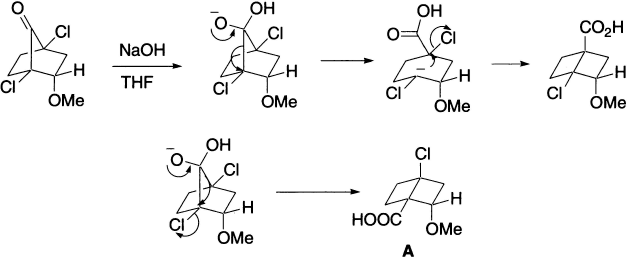
An interesting example is the conversion of the cyclobutyl into the cyclopropyl system, for this must apparently involve the highly strained bicyclobutane system.
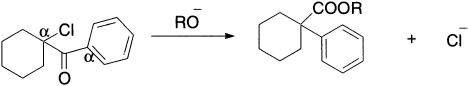
The cyclopropanone mechanism for the Favorskii rearrangement is obviously excluded for ketones such as 1-benzoylcyclohexyl chloride, in which the nonhalogenated α-carbon bears no hydrogen atom. Nevertheless, this ketone is found to rearrange with ease to ester. It thus appears that there are at least two distinct paths by which the Favorskii rearrangement may proceed, and that a mechanism analogous to the benzilic acid rearrangement (known as ‘semibenzilic’ rearrangement) may operate when cyclopropanone mechanism may not. If the reaction were concerted, elimination of the other chlorine would be just as favourable and the isomeric product A would be formed.
Leave a Reply