In 1959 Winstein introduced the term ‘homoaromatic’ to describe compounds that display aromaticity despite one or more saturated linkages interrupting the formal cyclic conjugation. Homoaromaticity is a term used to describe systems in which a stabilized cyclic conjugated system is formed by bypassing one saturated atom. The saturated unit is generally a CH2 group, but can be a larger alkyl residue or even a heteroatomic moiety. The resulting stabilization would, in general, be expected to be reduced because of poorer overlap of the orbitals. The properties of several such cationic species, however, suggest that substantial stabilization does exist.
Homoaromaticity is well established in cationic systems where delocalization of charge provides an additional driving force for homoaromaticity. The homocyclopropenium cation (I) is the simplest homoaromatic, and its derivative (II) was the first system proposed to be homoaromatic. These homoaromatic cations adopt a puckered geometry with a relatively short non-bonded 1,3- distance (~1.8 Å, consistent with homoaromaticity), a high barrier to inversion (Ia Ib, ~ 8 kcal/mol) through a planar non-aromatic transition state. The size of the barrier to inversion is taken as an indicator of the degree of homoaromaticity.
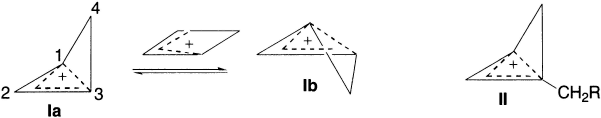
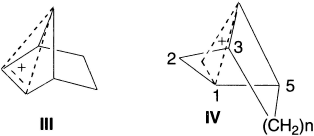
Roberts et. al. first proposed the enhanced stability of the bishomocyclopropenyl cation (III); numerous 3,5-bridged derivatives (IV) were prepared and fully characterized as homoaromatic, both by experiment and theory.
The homotropylium cation (V) and its numerous derivatives are the most extensively studied and well-established homoaromatics. V is the archetype no-bond homoaromatic species. It adopts a boat-shaped conformation with the seven-member ring not greatly deviating from planarity. There is no bond-critical point linking C1-C7, but there is a significant build up of electron density between these carbon atoms, which mediates effective (through-space) delocalization. The positive charge is evenly distributed over the seven-member ring, as evidenced by calculation, and 13C NMR chemical shifts for C1-C7. All estimates indicate that the homotropylium cation is more stable than the reference compound used and, therefore, support its homoaromaticity.
A significant feature of the NMR spectrum of this cation is that both C8 protons are shielded and exhibit sharply different chemical shifts; that is, the 8-endo proton resonates at higher field than tetramethylsilane. The NMR chemical shift difference between the 8-endo and 8-exo protons is large (5.86 ppm). Molecular orbital calculations and the effects of electron correlation indicate that the homoconjugated structure is a good description of the cation and find that there is a strong aromatic ring current.
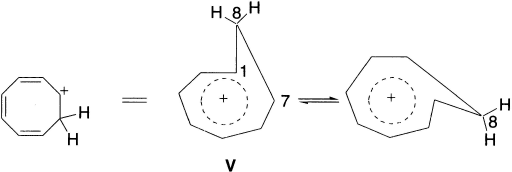
The temperature-dependent NMR spectrum of the ion can be analyzed to show that there is a barrier (8.4 kcal/mol) for the ring flip that interchanges the two hydrogens of the methylene group. The 13C-NMR chemical shift is also compatible with the homoaromatic structure. Molecular orbital calculations are successful in reproducing the structural and spectroscopic characteristics of the cation and are consistent with a homoaromatic structure.
Childs, Cremer and Elia in 1995 considered that there may be examples of anionic homoaromatics. The bicyclo[3.2.1]octadienyl anion (VI) is the potential homoaromatic anion.
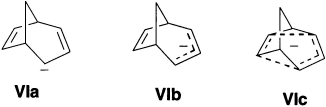
The existence of stabilizing homoconjugation in anions has been more difficult to establish. Much of the discussion has revolved about anion VI. This species was proposed to have aromatic character on the basis of the large upfield shift of the CH2 group that would lie in the shielding region generated by a diamagnetic ring current. The 13C NMR spectrum can also be interpreted in terms of homoaromaticity. Both gas-phase and solution measurements suggest that the parent hydrocarbon is more acidic than would be anticipated if there were no special stabilization of the anion. An X-ray crystal structure of the lithium salt has been done. The lithium is not symmetrically disposed toward the anion, but is closer to one carbon of the allyl system. There is no indication of flattening of the homoconjugated atoms, and the C(6)-C(7) bond distance is in the normal double-bond range (1.354 Å). Recently, many anionic compounds have been fully characterized as homoaromatic.
Leave a Reply