The meaning of the word aromaticity has evolved, as understanding of the special properties of benzene and other aromatic molecules has deepened. Originally, aromaticity was associated with a special chemical reactivity. The aromatic hydrocarbons were considered to be those unsaturated systems that underwent substitution reactions in preference to addition. Later, the idea of special stability became more important. Benzene can be shown to be much lower in enthalpy than predicted by summation of the normal bond energies for the C=C, C–C, and C–H bonds in the Kekule representation of benzene. Aromaticity is now generally associated with this property of special stability of certain completely conjugated cyclic molecules. A major contribution to the stability of aromatic systems results from the delocalization of electrons in these molecules.
Aromaticity is usually described in molecular orbital terminology. Cyclic structures that have a particularly stable arrangement of occupied p molecular orbitals are called aromatic. A simple expression of the relationship between a molecular orbital description of structure and aromaticity is known as the Hückel rule. The German chemist Erich Hückel was the first to recognize, in 1931, that an aromatic compound must have an odd number of pairs of π electrons. It is derived from the Huckel molecular orbital (HMO) theory and states that planar monocyclic completely conjugated hydrocarbons will be aromatic when the ring contains 4n + 2π electrons. The Huckel rule can be applied to charged as well as neutral systems.
Simple Huckel calculations on benzene place all the π electrons in bonding molecular orbitals. The π-electron energy of benzene is calculated by summing the energies of the six π electrons, which is 6α + 8β, lower by 2β than the value of 6α + 6β for three isolated double bonds. Thus, the HMO method predicts a special stabilization for benzene.
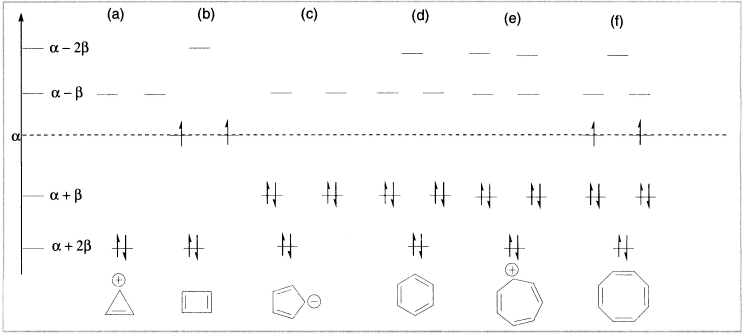
Figure 12.1 Energy levels of HMOs of cyclic π system
The lowest-energy α+2β bonding orbital has no nodes, and the two degenerate α+β bonding orbitals each have one node. The degenerate α–β antibonding orbitals have two nodes, and the α–2β orbital, three nodes. The pattern of two half-filled degenerate levels persists for larger rings containing 4n + 2 electrons. In contrast, all 4n + 2 systems are predicted to have all electrons paired in bonding molecular orbitals with net stabilization relative to isolated double bonds. This pattern provides the theoretical basis of the Huckel rule.
Attempts to describe just how stable a given aromatic molecule is in terms of simple HMO calculations have centred on the delocalization energy. The total π-electron energy of a molecule is expressed in terms of the energy parameters α (the coulomb integral) and β (the resonance integral) that arise in HMO calculations. This energy value can be compared to that for a hypothetical localized version of the same molecule. The HMO energy for the π electrons of benzene is 6α+8β. The same quantity for the hypothetical localized model cyclohexatriene is 6α+6β, the sum of three isolated C=C bonds. The difference of 2β is called the delocalization energy or resonance energy. Although this quantity is often useful for comparing related systems, it is not a measurable physical quantity; rather, it is obtained by comparing a real molecule and a hypothetical one. Most estimates of the stabilization of benzene are in the range of 20–40 kcal/mol and depend on the choice of properties assigned to the hypothetical cyclohexatriene reference point.
There have been two general approaches to determining the amount of stabilization that results from aromatic delocalization. One is to use experimental thermodynamic measurements. Bond energies are nearly additive when there are no special interactions between the various bond types. Thus, it is possible to calculate such quantities as the heat of combustion or heat of hydrogenation of ‘cyclohexatriene’ by assuming that it is a compound with no interaction between the conjugated double bonds. For example, a very simple calculation of the heat of hydrogenation for cyclohexatriene would be to multiply the heat of hydrogenation of cyclohexene by 3, that is, 3 × 28.6 = 85.8 kcal/mol. The actual heat of hydrogenation of benzene is 49.8 kcal/mol, suggesting a total stabilization or delocalization energy of 36.0 kcal/mol. There are other, more elaborate, ways of approximating the thermodynamic properties of the hypothetical cyclohexatriene. The difference between the calculated and corresponding measured thermodynamic property of benzene is taken to be the aromatic stabilization. For benzene, the values obtained are usually around 30 kcal/mol, but the aromatic stabilization cannot be determined in an absolute sense because these values are established by the properties assigned to the cyclohexatriene model.
The second general approach to estimating aromatic stabilization is to use molecular orbital methods. This has already been illustrated by the discussion of benzene according to simple HMO theory, which assigns the stabilization energy a value of 2β units. More advanced molecular orbital methods can assign the stabilization energy in a more quantitative way. The most successful method is to perform calculations on the aromatic compound and on a linear, conjugated polyene containing the same number of double bonds. This method assigns a resonance stabilization of zero to the polyene, even though it is known by thermodynamic criteria that conjugated polyenes do have some stabilization relative to isomeric compounds with isolated double bonds. Using this definition, semi empirical molecular orbital calculations assign a value of about 20 kcal/mol to the resonance energy of benzene, relative to 1,3,5-hexatriene. The use of polyenes as reference compounds has proven to give better agreement with experimental trends in stability than comparison with the sums of isolated double bonds.
Both thermochemical and molecular orbital approaches agree that benzene is an especially stable molecule and are reasonably consistent with one another in the stabilization energy that is assigned. It is very significant that molecular orbital calculations also show a destabilization of certain conjugated cyclic polyenes, cyclobutadiene in particular. The instability of cyclobutadiene has precluded any thermochemical evaluation of the extent of destabilization. Compounds that are destabilized relative to conjugated noncyclic polyene models are called antiaromatic.
There are also physical measurements that can give evidence of aromaticity. The determination of the bond lengths in benzene by electron diffraction is a classic example of use of the bond-length criterion of aromaticity. Spectroscopic methods or X-ray diffraction can also provide bond-length data. Aromatic hydrocarbons show carbon-carbon bond lengths in the range 1.38–1.40 Å, and the bond lengths are quite uniform around the ring. In contrast, localized polyenes show alternation between typical sp3–sp3 single bond and sp2–sp2 double bond lengths along the conjugated chain. The uniformity of bond lengths has been developed as a criterion of aromaticity.
NMR spectroscopy also provides an experimental tool capable of assessing aromaticity. Aromatic compounds exhibit a diamagnetic ring current. Qualitatively, this ring current can be viewed as the migration of the delocalized electrons in the aromatic system under the influence of the magnetic field in an NMR spectrometer. The ring current effect is responsible for a large magnetic anisotropy in aromatic compounds. The induced ring current gives rise to a local magnetic field that is opposed to the direction of the applied magnetic field. Nuclei in a cone above or below the plane of an aromatic ring are shielded by the induced field and appear at relatively high field in the NMR spectrum, whereas nuclei in the plane of the ring, that is, the atoms bound directly to the ring occur at downfield positions. Antiaromatic compounds show opposite effects. The occurrence of these chemical shift phenomena is evidence for aromaticity. The chemical shift phenomena can be treated on a quantitative basis by quantum-mechanical calculation of the chemical shift at the centre of the ring. The value of the chemical shift at a point in the centre of the ring can be calculated. These values are referred to as the nucleus-independent chemical shift (NICS). These values show excellent correlation with other manifestations of aromaticity. Benzenoid hydrocarbons such as benzene, naphthalene and anthracene show values of about −9 to −10 ppm. Heteroaromatic five-member rings show slightly more negative values (pyrrole, −15.1; thiophene, −13.6; furan, −12.3). The values for aromatic ions such as cyclopentadienide (−14.3) and cycloheptatrienylium (−7.6) are also negative. Those for antiaromatic species, including cyclobutadiene (+27.6) and borole (+17.5), are positive. Saturated compounds such as cyclohexane have values near zero.
Another property associated with aromaticity is magnetic susceptibility. Magnetic susceptibility is determined by measuring the force exerted on the sample by a magnetic field. Magnetic susceptibility can also be determined using an NMR spectrometer. It is noted that aromatic compounds have enhanced magnetic susceptibility, relative to values predicted on the basis of the localized structural components. Magnetic susceptibility can also be calculated by computational methods.
It has been argued that there are two fundamental aspects of aromaticity, one reflecting structure and energy, and the other, magnetic properties and electron mobility. Parameters of aromaticity such as bond length and stabilization appear to be largely independent of the magnetic criteria, such as diamagnetic ring current. However, there is a correlation between the two kinds of measurements. The more stabilized compounds exhibit the greatest magnetic susceptibility. Aromaticity is thus best conceived as a single property resulting from cyclic delocalization that results in both stabilization and the magnetic phenomena associated with electron mobility.
The experimental resonance energies of polynuclear aromatic compounds such as naphthalene, anthracene and phenanthrene are found to be 61.0, 83.5 and 91.3 kcal/mol, respectively. Naphthalene is like benzene and behaves almost similarly. Phenanthrene, however, undergoes electrophilic addition with bromine, suggesting that greater resonance energy does not necessarily result in more aromatic behaviour.
Leave a Reply