There is evidence that aromatic segments can exist as part of larger conjugated units, resulting in an aromatic segment in conjugation with a ‘localized’ double bond. For example, in acenaphthylene, the double bond in the five-member ring is both structurally and chemically similar to a normal localized double bond. The resonance energy 0.57β is slightly less than that for naphthalene (0.59β). The additional double bond of acenaphthylene has only a small effect on the stability of the conjugated system. The molecular structure determined at 80 K by neutron diffraction shows bond lengths for the aromatic portion that are very similar to those of naphthalene. The double bond is somewhat longer than a normal double bond, but this may reflect the strain imposed by the naphthalene framework on the double bond.
The predictions of relative stability obtained by the various approaches diverge more widely when nonbenzenoid systems are considered. The simple Huckel method using total π delocalization energies relative to isolated double-bond reference energy (α + β) fails. This approach predicts stabilization of the same order of magnitude for such unstable systems as pentalene and fulvalene, as it does for much more stable aromatics. The HMO, RE and SCF-MO methods, which use polyene reference energies, do much better. All show drastically reduced stabilization for such systems and, in fact, indicate destabilization of systems such as butalene and pentalene.
It is of interest to consider at this point some of the specific molecules and compare their chemical properties with the calculated stabilization energies. Benzocyclobutadiene has been generated in a number of ways, including dehalogenation of dibromobenzocyclobutene. The compound is highly reactive, dimerizing or polymerizing readily. Benzocyclobutadiene is very reactive as a dienophile in the Diels-Alder reaction. Generation of benzocyclobutadiene by fluoride-induced elimination has permitted the NMR spectrum to be observed under flow conditions.
All the peaks are somewhat upfield of the aromatic region, suggesting polyene character. This structure would also be consistent with the observed reactivity since the polyene has a quinodimethane structure. The implication of a non-aromatic structure is that the combination of ring strain and the anti-aromaticity associated with the four-member ring results in a localized system.
Azulene is one of the few nonbenzenoid hydrocarbons that appear to have appreciable aromatic stabilization. There is some divergence on this point between the SCF-MO and HMO results. The latter estimates a resonance energy about half that for the isomeric naphthalene, whereas the SCF-MO method assigns a resonance energy that is only about one-seventh that of naphthalene. Naphthalene is more stable than azulene by about 38.5 kcal/mol. Molecular mechanics calculations attribute about 12.5 kcal/mol of this difference to strain and about 26 kcal/mol to greater resonance stabilization of naphthalene. Based on heats of hydrogenation, the stabilization energy of azulene is about 16 kcal/mol. The parent hydrocarbon and many of its derivatives are well-characterized compounds with considerable stability. The structure of azulene has been determined by both X-ray crystallography and electron diffraction measurements. The peripheral bond lengths are in the aromatic range and show no regular alternation. The bond shared by the two rings is significantly longer, indicating that it has predominantly single-bond character. Theoretical calculations indicate that the molecule has C2v symmetry, indicating delocalization of the π electrons.
An interesting structural question involves the contribution of a dipolar structure, which pictures the molecule as the fusion of a cyclopentadienide anion and a cycloheptatrienyl cation.
Azulene does have an appreciable dipole moment (0.8 D). The essentially single-bond nature of the shared bond indicates, however, that the conjugation is principally around the periphery of the molecule. Several molecular orbital calculations have been applied to azulene. Calculations, which include electron correlation effects, give a delocalized π system as the minimum-energy structure. In contrast to the significant resonance stabilization of azulene, pentalene and heptalene are indicated to be destabilized relative to a reference polyene.
Preparation of pentalene is followed by immediate dimerization. Low-temperature photolysis produces a new species believed to be pentalene, but the compound reverts to dimer at 100°C. The matrix-isolated monomer has been characterized spectroscopically. The results are in accord with the predicted lack of stabilization.
Heptalene readily polymerizes and is sensitive to oxygen. The NMR spectrum does not indicate the presence of an aromatic ring current. The conjugate acid of heptalene, however, is very stable (even at pH 7 in aqueous solution), reflecting the stability of the cation, which is a substituted tropylium ion.
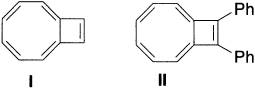
Another structure with a 10-π-electron conjugated system is bicyclo[6.2.0]deca-1,3,5,7,9-pentaene (I), which is neither aromatic nor anti-aromatic. The crystal structure of the 9,10-diphenyl derivative (II) shows the conjugated system to be nearly planar. There is significant bond alternation; however, the bond at the ring fusion is quite long (1.539 Å). A molecular mechanics calculation on this molecule that included an SCF-MO treatment of the planar-conjugated system found the molecule to be slightly destabilized (4kcal/mol) relative to a polyene reference.
The possibility of extra stabilization in conjugated systems that have conjugated components exocyclic to the ring has also been examined. The substituents complete conjugated rings but are not part of the cyclic system. Cyclopropenes and cyclopentadienes with exocyclic double bonds provide the possibility of dipolar resonance structures that suggest aromatic character in the cyclic structure.
For methylenecyclopropene, a microwave structure determination has established bond lengths, which show the strong alternation anticipated for a localized structure. The molecule does have a significant dipole moment (1.90 D), implying a contribution from the dipolar resonance structure. Fulvenes are cyclic cross-conjugated molecules with an odd number of carbon atoms in the ring. According to the size of the ring skeleton, they are named tri-, penta-, hepta- and nona-fulvenes. The molecular geometry of dimethylfulvene has been examined by electron diffraction methods. Strong bond-length alternation indicative of a localized structure is found. The fulvalene systems are not predicted to be aromatic by any of the theoretical estimates of stability. Even simple resonance considerations would suggest polyene behaviour, since only dipolar resonance structures can be drawn in addition to the single non-polar structure. Trifulvalene (cyclopropenylidenecyclopropene) has not been isolated. A substantial number of pentafulvalene derivatives have been prepared. The chemical properties of these molecules are those of reactive polyenes. The NMR spectrum of pentafulvalene is characteristic of a localized system. Heptafulvalene is a well-characterized compound with the properties expected for a polyene.
Because the five-member ring is a substituted cyclopentadienide anion in some dipolar resonance structures, it might be expected that exocyclic groups that could strongly stabilize a positive charge would lead to a larger contribution from dipolar structures and enhanced stability. The stability of such dipolar systems depends on the balance between the increase in energy required to separate unlike charges and the aromaticity associated with Huckel 4n + 2 systems. Phenyl-substituted analogs are known and the large measured dipole moments suggest considerable charge separation. Some alkyl derivatives have been prepared. Their chemical behaviour is that of highly reactive polyenes. One interesting property that is revealed by the NMR spectra is a reduced barrier to rotation about the double bond between the two rings. This property suggests that rotation about this bond takes place easily through a transition state in which the two charged aromatic rings are twisted out of conjugation.
The hydrocarbon phenalene is the precursor of both a highly stabilized anion and a highly stabilized cation. The single orbital at the nonbonding level is the LUMO in the cation and the HOMO in the anion. The stabilization energy calculated for both would be the same and is 0.41β by the HMO comparison. The pK for conversion of phenalene to its anion is 19. Several methods for generating the phenatenyl cation have been developed. Because the centre carbon is part of the conjugated system, the Huckel rule, which applies only to monocyclic conjugated systems, cannot be applied to just the peripheral conjugation.
In general, the HMO and SCF methods both appear able to make reasonably accurate predictions about the stabilization in conjugated molecules. The stabilization is general for benzenoid compounds but quite restricted in nonbenzenoid systems. Because the HMO method of estimating stability is based on the ideas of HMO theory, its general success vindicates the ability of this very simplified molecular orbital approach to provide insight into the structural nature of the annulenes and other conjugated polyenes.
Leave a Reply