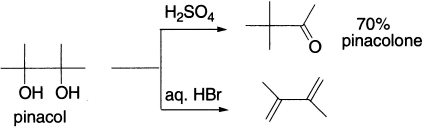
This is an acid catalyzed 1,2–migration of a diol to an oxo derivative. When vic–diols (glycols) are treated with acids, they can be rearranged to give aldehydes or ketones. The reaction gets its name from the typical compound pinacol Me2C(OH)C(OH)Me2, which is rearranged to pinacolone Me3CCOCH3. The rearrangement has been carried out with alkyl, aryl, hydrogen and even ethoxy carbonyl (COOEt) as migrating groups. Di–tert–glycols rearrange in the presence of acid to give α–tert–ketones. The reaction takes place in dilute or moderately concentrated acid at higher temperature.
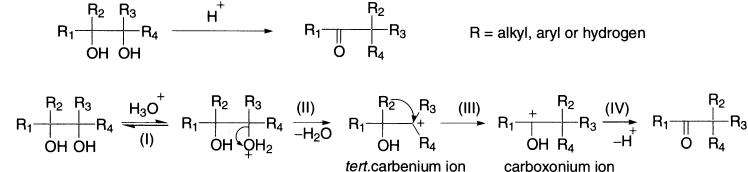
The first step is the reversible protonation of the relatively basic hydroxyl group. This was confirmed by a demonstration that threo– and erythro-1,2-diphenyl-1-p-tolylethylene glycols interconverted in dilute acid more rapidly than they rearranged. The second step is the dehydration, that is, formation of tert-carbenium ion. The carbenium ion rearranges in the third step, into a more stable carboxonium ion via a [1,2]- shift. In the last step the carboxonium ion is deprotonated and the product ketone is obtained. The driving force for this process is the stabilization of the new carboxonium ion by the loss of a proton from the hydroxyl group. The OH group, which forms the most stable carbenium ion, is protonated preferentially and a subsequent intramolecular migration yields the ketone. The reaction is unimolecular with regard to the concentration of the diol.
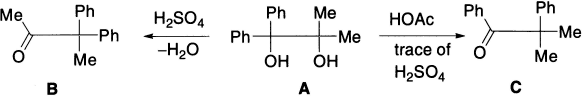
Glycols in which the four R groups are not identical can give rise to more than one product depending on which group migrates, which depends on the reaction conditions as well as on the nature of the substrate. Thus the action of cold, concentrated H2SO4 on A produces mainly 3,3–diphenyl–2–butanone B (methyl migration), while treatment of A with AcOH containing a trace of H2SO4 gives mainly C (phenyl migration).
Of the two OH groups, the one that forms the more stable carbocation is protonated preferentially. This factor takes precedence over the migratory aptitude factor.
The ease with which any particular group will undergo nucleophilic 1,2-shift is known as its migratory aptitude. There are two ways in which relative migratory aptitudes may be determined. One way involves the comparison of relative rates of rearrangement in a homologous series of substrates, each possessing just one type of migrating group, under exactly the same reaction condition. An alternative method analyzes the product mixtures obtained from a substrate containing two different competing substituents at the migrating origin. A wide range of other factors, such as steric and conformational effects also play an important role in determining whether a particular migration is favoured or not.
The migratory aptitude of the groups decreases in the order aryl > alkyl > hydrogen and amongst the aryl groups p-anisyl > p-tolyl > m-tolyl > m-anisyl > phenyl > p-chlorophenyl > o-anisyl > o-tolyl. The migratory aptitude increases as the aromatic nucleus is made more electron-rich, because the migrating group leaves with its electron-pair and makes a nucleophilic attack at the migration terminus. Electron-withdrawing substituents disfavour delocalization of positive charge to the benzene ring and exert the opposite effect. In unsymmetrical systems, the stability of the intermediate carbenium ion governs the migration of the group and this factor takes precedence over the migratory aptitude factor.

Of course, the way to carry out a direct comparison of relative migratory aptitudes of methyl versus phenyl in the pinacol rearrangement is to use 2,3-diphenylbutan-2,3-diol, as loss of either hydroxyl group generates the same carbocation.
In a crossover experiment, a mixture of di-tert-glycols 1,1-dimethyl-2,2-diphenyl glycol A and 1,1-diethyl-2,2-diphenyl glycol E was rearranged under acidic conditions. The reaction products were the ketones, 3,3-diphenyl-2-butanone C and 4,4-diphenyl-3-haxanone G.
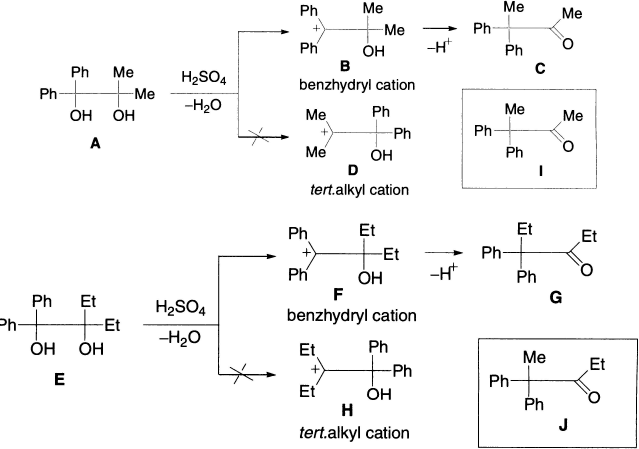
The absence of the crossover products I and J proves the intramolecular nature of the pinacol rearrangement.
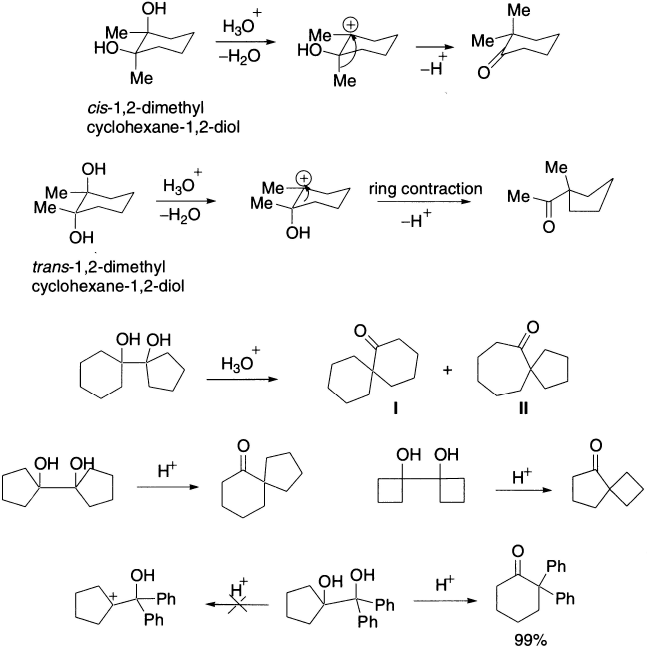
This rearrangement follows a definite stereochemical course in that the migrating group enters trans to the departing group, that is, reaction occurs in an anti-manner. This has important consequences in alicyclic systems. For example, cis-1,2-dimethyl-cyclohexane-1,2-diol undergoes a methyl shift to give 2,2-dimethylcyclohaxanone, whereas the trans-isomer undergoes ring contraction to give 1-acetyl-1-methyl cyclopentane. For cyclic diols, the rearrangement results in the contraction or expansion of the ring system, for example, cyclopentylcyclohexane-1,2-diol yields two products, the I being the major one.
Further evidence for carbenium ion formation in the pinacol rearrangement has been obtained by oxygen–exchange experiments. Partial rearrangement of pinacol to pinacolone was carried out in acidic solutions containing H2O18.
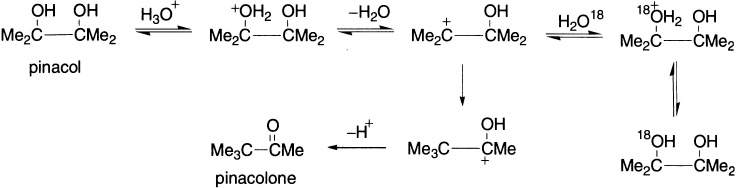
It was found that the recovered pinacol contained isotopically labelled oxygen, which established that formation of carbenium ion from pinacol is a reversible process. In this connection, it has been found that under similar experimental conditions, mixtures of pinacol and pinacolone are formed from different precursors.
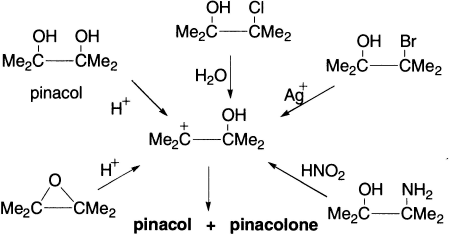
ß–aminoalcohols, Me2C(OH)C(NH2)Me2, which rearrange on treatment with nitrous acid; iodohydrins, Me2C(OH)C(I)Me2, for which the reagent is Hg2O or AgNO3; ß–hydroxyalkyl selenides, R1R2C(OH)C(SeR5)R3R4 and allylic alcohols may be rearranged with strong acids.
Leave a Reply