It has been observed that alkyl halides tend to proceed through E2 mechanism and protonated alcohols through E1. The vast majority of the two classes of eliminations use one of these two types of starting materials. Since the leaving group is involved in the rate-determining step of both, E1 and E2, in general, any good leaving group will lead to fast elimination. The leaving group’s ability parallels the stability of anions. For example, a comparison of the facility of ionization within a series of tert-butyl halides reveals that the rate decreases with the increasing strength of the carbon-halogen bond. R-I (51 kcal/mol) < R-Br (68 kcal/mol) < R-Cl (82 kcal/mol) < R-F (106 kcal/mol).
In the first example, a stabilized cation cannot be formed (so E1 is impossible), but a strong base is used, allowing E2. In the second, a stabilized tertiary cation could be formed (so either E1 or E2 might occur), but no strong base is present, so the mechanism must be E1.
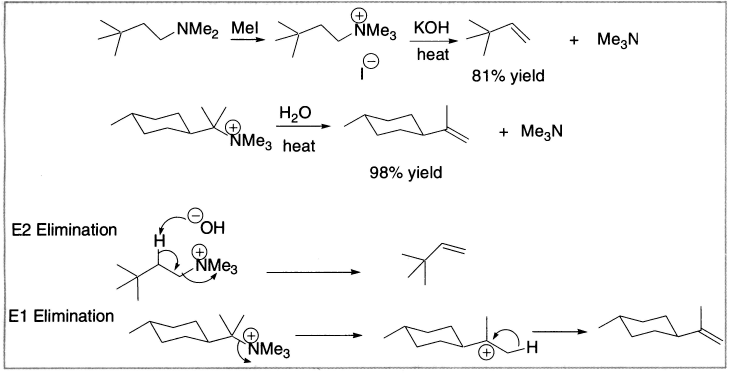
Figure 4.3 Elimination from quaternary ammonium salts
We have just seen that hydroxyl groups can be turned into good leaving groups in acid, but this is only useful for substrates that can react by E1 elimination. The hydroxyl group is never a leaving group in E2 eliminations, since they have to be done in base.
OH− is never a leaving group in an E2 reaction
For primary and secondary alcohols, the hydroxyl is best made into a leaving group for elimination reactions by sulphonylation, with toluene-para-sulphonyl chloride (tosyl chloride, TsCl) or methanesulphonyl chloride (mesyl chloride, MeSO2Cl or MsCl). Toluenesulphonate esters (tosylates) can be made from alcohols (with TsCl, pyridine). We have already met tosylates because they are good electrophiles for substitution reactions with nonbasic nucleophiles. With strong bases such as t-BuOK, NaOEt, DBU, or DBN, they undergo very efficient elimination reactions. Here are two examples.

Figure 4.4 E2 Elimination of tosylates
Methanesulphonyl chloride may be a new reagent to you. In the presence of base (usually triethylamine, Et3N) it reacts with alcohols to give methane-sulphonate esters, but the mechanism differs from the mechanism with TsCl. The first step is an elimination of HCl from the sulphonyl chloride (this cannot happen with TsCl, because there are no available protons) to give a sulphene. The sulphene is highly electrophilic at sulphur, and will react with any alcohol (including tertiary alcohols, which react very slowly with TsCl). Here are the two mechanisms compared.
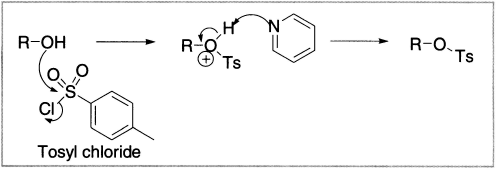
Figure 4.5 Formation of toluenesulphonates (tosylates)
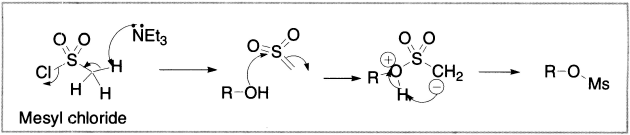
Figure 4.6 Formation of methanesulphonates (mesylates)
Methanesulphonyl esters (or mesylates) can be eliminated using DBU or DBN, but a good way of using MsCl to convert alcohols to alkenes is to do the mesylation and elimination steps in one go, using the same base (Et3N) for both.
Stereoselective E1 Reactions
For some eliminations, only one product is possible. For others, there may be a choice of two (or more) alkene products that differ either in the location or stereochemistry of the double bond. We shall now move on to discuss the factors that control the stereochemistry (geometry) and regiochemistry (that is, where the double bond is) of the alkenes, starting with E1 reactions.
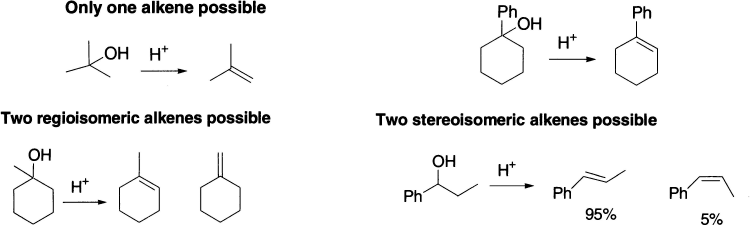
For steric reasons, E-alkenes (and transition states leading to E-alkenes) are usually lower in energy than Z-alkenes (and the transition state leading to them) because the substitution can get farther apart from one another. A reaction that can choose which product it forms is therefore likely to favour the formation of E-alkenes. For alkenes formed by E1 elimination, this is exactly what happens: the less hindered E-alkene is favoured.
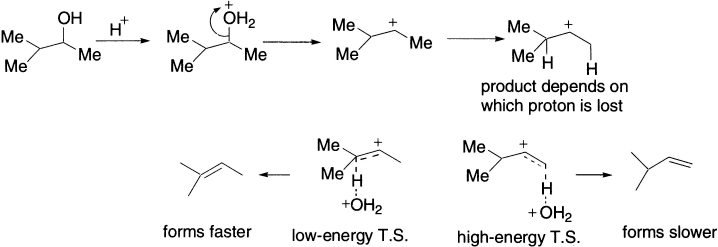
The geometry of the product is determined at the moment that the proton is lost from the intermediate carbocation. The new π bond can only form if the vacant p orbital of the carbocation and the breaking C-H bond are aligned parallel. There are two possible conformations of the carbocation with parallel orientations, but one is more stable than the other because it suffers less steric hindrance. The same is true of the transition states on the route to the alkenes-the one leading to the E-alkene is lower in energy and more E-alkene than Z-alkene is formed. The process is stereoselective, because the reaction chooses to form predominantly one of two possible stereoisomeric products.
Regioselective E1 Reactions
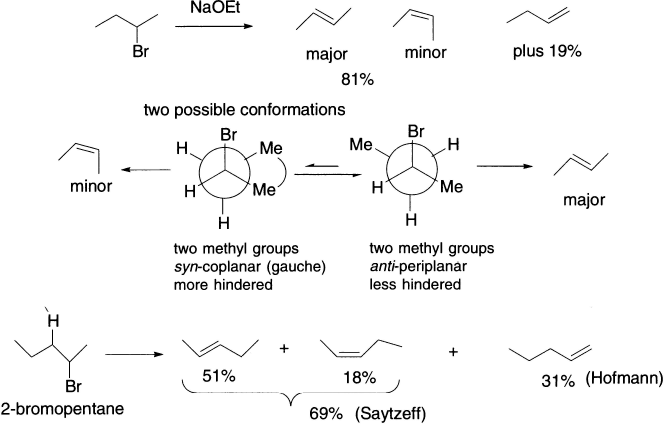
E1 eliminations can give more than one regioisomeric alkene. The major product is the alkene that has more substituents (Saytzeff rule), because this alkene is the more stable of the two possible products. Here is an example.

An exception is found when the product of Saytzeff elimination is destabilized by serious steric interaction for example, between a methyl group and t-butyl group.
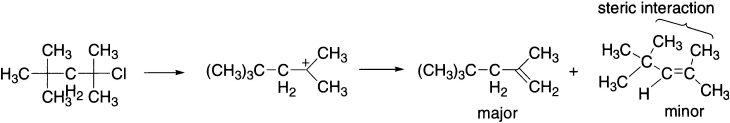
The reason for this is related to the reason why more substituted carbocations are more stable. It is well established that the carbocation is stabilized when its empty p-orbital can interact with the filled orbital of parallel C–H and C–C bonds. The same is true of the π system of the double bond-it is stabilized when the empty π* antibonding orbital can interact with the filled orbital of parallel C–H and C–C bonds. The more C–C or C–H bonds there are, the more stable the alkene.
The more substituted alkene is more stable, but this does not necessarily explain why it is the one that forms faster. To do that, we should look at the transition states leading to the two alkenes. Both form from the same carbocation, but which one we get depends on which proton is lost. Removal of the proton on the right leads to a transition state in which there is a monosubstituted double bond partly formed. Removal of the proton on the left leads to a partial double bond that is trisubstituted. This is more stable–the transition state is lower in energy, and the more substituted alkene forms faster.
Although E1 reactions show some stereo- and regioselectivity, the level of selectivity in E2 reactions can be much higher because of the more stringent demands on the transition state for E2 elimination.
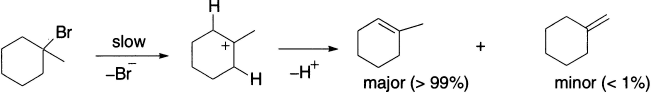
Loss of HBr from 1-bromo-1-methylcyclohexane leads to two regioisomeric alkenes. The major product, 1-methylcyclohexene, in which the double bond is in the ring (endocyclic) is the most stable isomer. On the other hand, methylenecyclohexane, in which the double bond is outside the ring (exocyclic), is the least stable isomer.
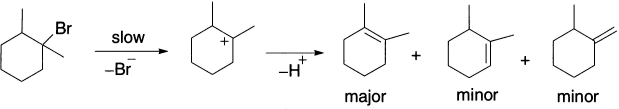
Another example is the elimination of HBr from 1-bromo-1,2-dimethyl-cyclohexane that leads to three regioisomeric alkenes.
Anti-periplanar Transition States of E2 Eliminations
In E2 elimination, the new π bond is formed by overlap of the C–H σ bond with C–X σ* antibonding orbital. The two orbitals have to lie in the same plane for the best overlap, and now there are two conformations that allow this. One has H and X syn-periplanar (that is, H and X are on the same side of the molecule; periplanar means both H and Y are in the same plane), the other, anti-periplanar (that is, H and X are on the opposite sides of the molecule but in the same plane). The anti-periplanar conformation is more stable because it is staggered (the syn-periplanar conformation is eclipsed) but, more importantly, only in the anti-periplanar conformation are the bonds (and therefore the orbitals) truly parallel.
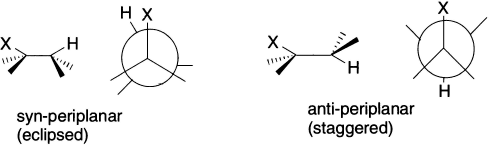
E2 eliminations therefore take place from anti-periplanar conformation. Thus, the dihedral angle between the proton and the leaving group in the anti-periplanar conformation is 180°. We shall see shortly how we know this to be the case, but first, we consider an E2 elimination that gives mainly one of two possible stereoisomers. 2-Bromobutane has two conformations with H and Br anti-periplanar, but the one that is less hindered leads to more of the product, and the E-alkene predominates. There is a choice of protons to be eliminated-the stereochemistry of the product results from which proton is anti-periplanar to the leaving group when the reaction takes place, and the reaction is stereoselective as a result. The transition state for syn-coplanar elimination is an eclipsed conformation of higher energy due to the eclipsing interactions and interference between the attacking base and the leaving group.
Stereospecific E2 Eliminations
In the next example, there is only one proton that can take part in the elimination. Now there is choice of anti-periplanar transition states. Whether the product is E or Z, the E2 reaction has only one course to follow, and the outcome depends on which diastereoisomer of the starting material is used. When the first diastereoisomer is drawn, with the proton and bromine anti-periplanar, as required, and in the plane of the page, the two phenyl groups have to lie one in front and one behind the plane of the paper. As the hydroxide attacks the C–H bond and eliminates Br −, this arrangement is preserved and the two phenyl groups end up trans (the alkene is E). This is perhaps easier to see in the Newman projection of the same conformation.
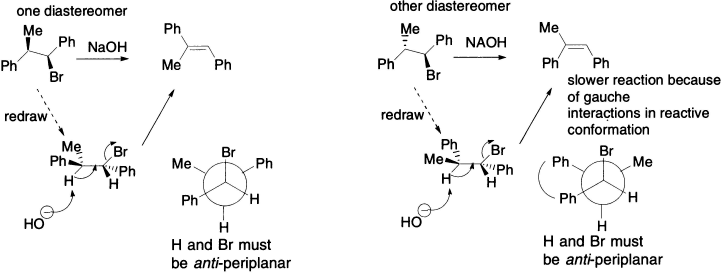
The second diastereoisomer forms the Z-alkene for the same reasons: the two phenyl groups are now on the same side of the H–C–C-Br plane in the reactive anti-periplanar conformation (again, this is clear in the Newman projection) and so they end up cis in the product. Each diastereoisomer gives different alkene geometry, and they do so at different rates. The first reaction is about ten times as fast as the second because, although this anti-periplanar conformation is the only reactive one, it is not necessarily the most stable. The Newman projection for the second reaction shows clearly that the two phenyl groups have to lie synclinal (gauche) to one another: the steric interaction between these large groups will mean that, at any time, a relatively small proportion of molecules will adopt the right conformation for elimination, slowing the process down.
meso-2,3-Dibromobutane undergoes elimination of two bromines with iodide ion to give trans-2-butene, while the (d,1)-pair gives cis-2-butene. This is because elimination can occur only via a conformation in which the two bromine atoms are in an anti-periplanar arrangement. In the case of (d,1)-isomer, two methyl groups are gauche placed, involving a less stable transition state compared to the meso-isomer that results in elimination that is slower by a factor of about two.
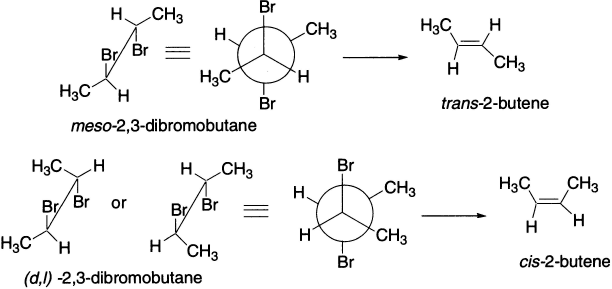
Reactions in which the stereochemistry of the product is determined by the stereochemistry of the starting material are called stereospecific.
Stereoselective or stereospecific
- Stereoselective reactions give one predominant product because the reaction pathway has a choice. Either the pathway of lower activation energy is preferred (kinetic control) or the more stable product (thermodynamic control).
- Stereospecific reactions lead to the production of a single isomer as a direct result of the mechanism of the reaction and the stereochemistry of the starting material. There is no choice. The reaction gives a different diastereomer of the product from each stereoisomer of the starting material.
Leave a Reply