Now that we have seen a few examples of elimination reactions, it is time to return to our discussion of the two mechanisms for elimination. To summarize what we have said so far:
- E1 describes an elimination reaction (E) in which the rate-determining step is unimolecular (1) and does not involve the base. The leaving group leaves in this step, and the proton is removed in a separate second step
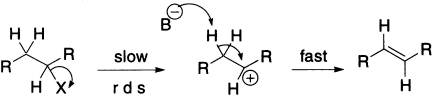
- E2 describes an elimination (E) that has a bimolecular (2) rate-determining step that must involve the base. The loss of the leaving group and removal of the proton is simultaneous.
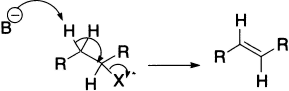
There are a number of factors that decide whether an elimination goes by an E1 or an E2 mechanism. One is immediately obvious from the rate equations: only the E2 is affected by the concentration of base, so at high base concentration, E2 is favoured. The rate of an E1 reaction is not even affected by what base is present, so E1 is just as likely with weak as with strong bases, while E2 goes faster with strong bases than weak ones: strong bases at any concentration will favour E2 over E1. If a strong base is being used for elimination, it is certainly an E2 reaction.
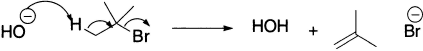
In a compound, which can eliminate to give different alkenes, the more highly substituted alkene is the major product. This generalization is known as the Saytzeff rule. Exceptions to the Saytzeff rule occur in two circumstances. First, when the proton to be removed is in the sterically more hindered environment, the use of a base whose basic centre is also sterically hindered can lead to the predominance of the less substituted alkene.
Second, when the substrates are quaternary ammonium, sulphonium or phosphonium salts, they give predominantly the less substituted alkene (Hofmann rule).
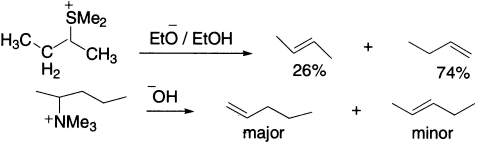
Iodide is the best and fluoride is the worst leaving group among halides. With fluorine, therefore, C-H bond-breaking prefers generating primary carbanion leading to Hofmann orientation.
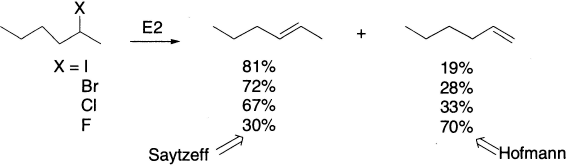
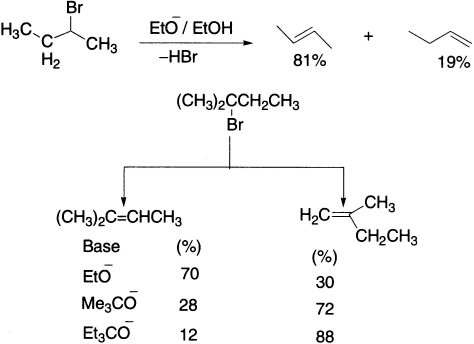
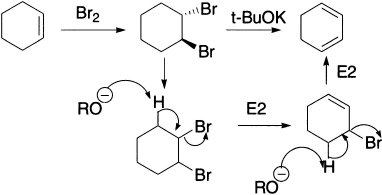
With less hindered alkyl halides, hydroxide would not be a good choice as a base for elimination because it is rather small and still very good at SN2 substitution. So, what are good alternatives? We have already mentioned that the bulky t-butoxide is ideal for promoting E2, as it is both bulky and a strong base (pKaH =18). The conversion of a dibromide to a diene with two successive E2 is a useful two-step conversion of an alkene to a diene.
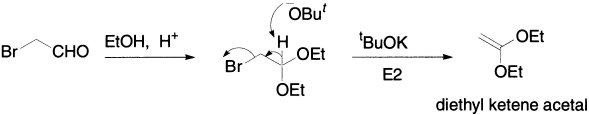
Unlike most acetals, diethylketene acetal cannot be formed directly from ketene, which is too unstable. Instead, the acetal is made by the usual method from bromoacetaldehyde, and then HBr is eliminated using t-BuOK.
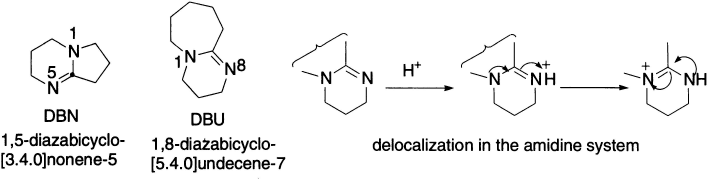
Among the most commonly used bases for converting alkyl halides to alkenes are two, 1,8-diazabicyclo-[5.4.0] undecene-7 (DBU) and 1,5-diazabicyclo-[3.4.0] nonene-5 (DBN). These two bases are amidines; delocalization of one nitrogen’s lone pair on to the other, and the resulting stabilization of the protonated amidinium ion, makes them particularly basic, with pKaHs of about 12.5. There is not much chance of getting those voluminous fused rings into tight corners, so they pick off the easy-to-reach protons rather than attacking carbon atoms in substitution reactions. DBU or DBN will generally eliminate HX from alkyl halides to give alkenes.
Substrate structure may allow E1 The elimination of t-BuBr illustrates something very important: the starting material is a tertiary alkyl halide (and would therefore substitute only by SN1); it can eliminate by either E2 (with strong bases) or E1 (with weak bases). The steric factors that disfavour SN2 at hindered centres do not exist for eliminations. Nonetheless, E1 can occur only with substrates that can ionize to give relatively stable carbocations, for example, tertiary, allylic or benzylic alkyl halides. For example, secondary alkyl halides may eliminate by E1, but primary alkyl halides only eliminate by E2 because the primary carbocation required for E1 would be too unstable. The chart on the facing page summarizes the types of substrates that can undergo E1; but remember that any of these substrates, under the appropriate conditions (in the presence of strong bases), may also undergo E2. Three alkyl halides that cannot eliminate by either mechanism, simply because they do not have any hydrogens to lose from carbon atoms adjacent to the leaving group, have also been included in this chart.
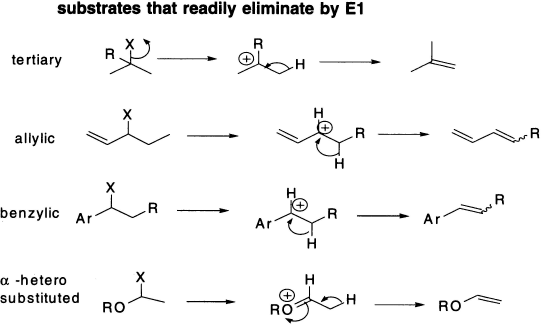
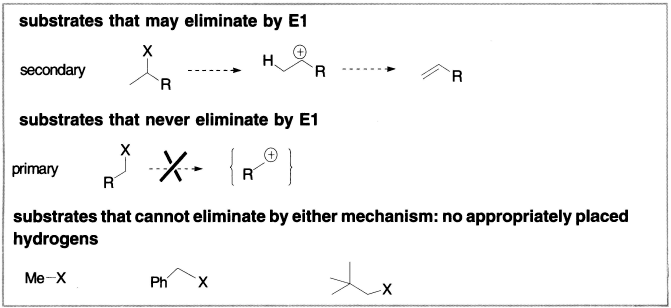
Figure 4.2 Elimination by substances
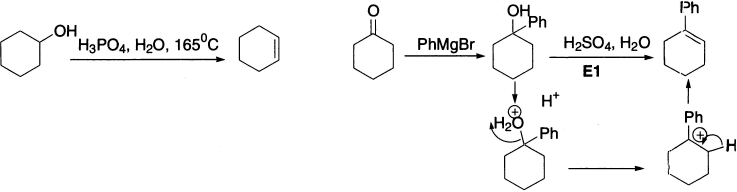
Polar solvents also favour E1 reactions because they stabilize the intermediate carbocation. E1 eliminations from alcohols in aqueous or alcoholic solutions are particularly common and very useful. An acid catalyst is used to promote loss of water, and in dilute H2SO4 or HCl the absence of good nucleophiles ensures that substitution does not compete. Under these conditions, the secondary alcohol cyclohexanol gives cyclohexene.
But the best E1 eliminations of all are with tertiary alcohols. The alcohols can be made by nucleophilic attack by an organometallic on a carbonyl compound. Note that the proton required in the first step is recovered in the last; the reaction requires only a catalytic amount of acid.
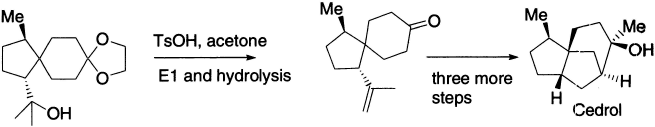
Cedrol is important in the perfumery industry; it has a cedar wood fragrance. Corey’s synthesis includes this step, the acid (toluenesulphonic acid) catalyzes both the E1 elimination and the hydrolysis of the acetal.
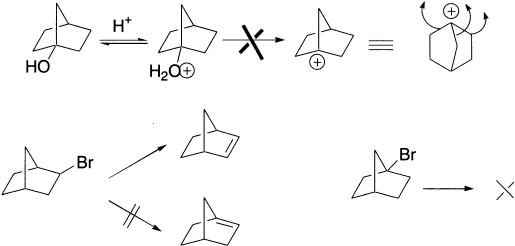
Bicyclic compound having a leaving group at the bridgehead will not undergo elimination by either an E1 or an E2 mechanism. We shall see shortly what the problem with E2 is, but, for E1, the hurdle to be overcome is the formation of a planar carbocation. The bicyclic structure prevents the bridgehead carbon becoming planar, although the cation would be tertiary; it is very high in energy and does not form. The impossibility of planar bridgehead carbons means that double bonds can never be formed to bridgehead carbons in bicyclic systems. This principle is known as Bredt’s rule.
Leave a Reply